
Accurately call genotypes on Illumina SNP arrays
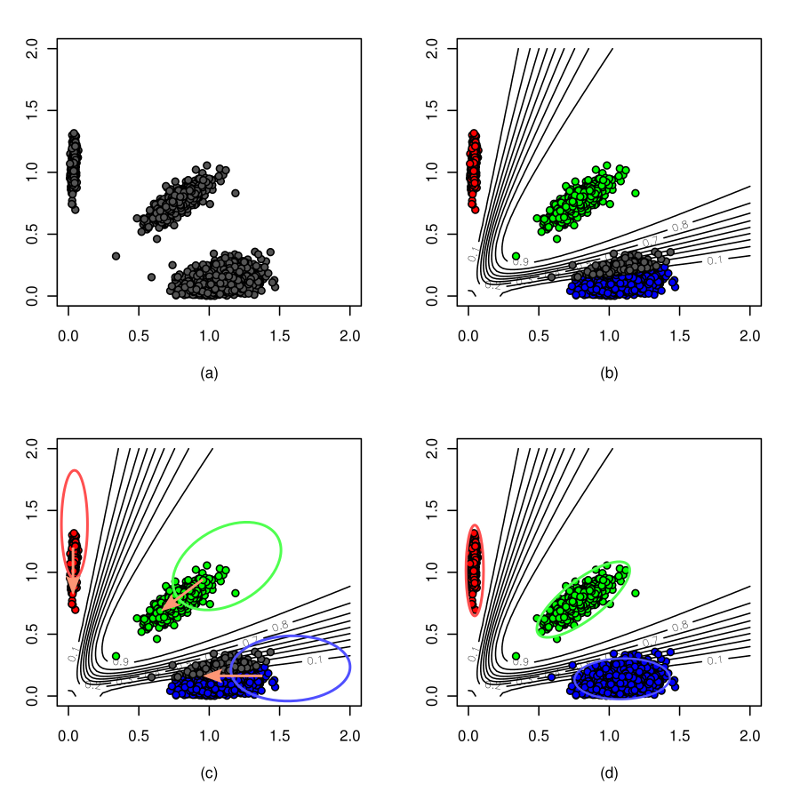
optiCall is designed to make accurate genotype calls across the minor allele frequency spectrum. Using intensity information from across multiple individuals and multiple SNPs when calling genotypes, allows it to call both rare and common variants accurately. For a full description of the method see here.
REQUIREMENTS:
optiCall includes all the libraries it needs, and requires nothing beyond the C++ standard. It compiles using g++ on unix/mac based systems.
CONTACT & SUPPORT:
If you have any queries, comments or feedback feel free to contact us at:
opticall@sanger.ac.uk
INSTALLATION:
uncompress the downloaded file by running:
tar -xvzf <filename>
which creates a folder of the same name as filename. cd into the optiCall folder:
cd <foldername>/opticall
then compile the optiCall code by running the make command:
make
after which the optiCall executable will appear, and you've successfully installed optiCall!
INPUT/OUPUT FORMATS:
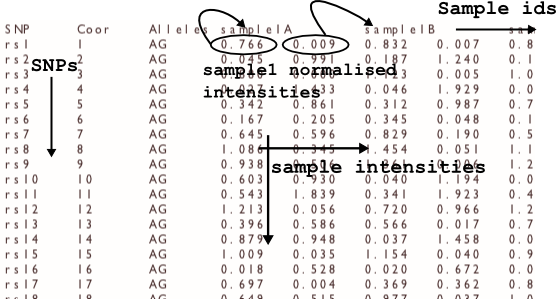
-in INPUT_FILE
optiCall reads in a file containing Illumina normalized intensities. The intensity input file is tab separated, with SNPs as rows, and samples as columns. So a line would be:
rsid <tab> rscoord <tab> allelesAB <tab> int1A <tab> int1B <tab> int2A <tab> int2B etc.
where int1A is the allele A intensity value for sample1, and id1B is the allele B intensity value for sample1. Any missing intensities should be input as NaN for both the A and B alleles.
The first line of the file should also be a header line of the form:
SNP <tab> Coor <tab> Alleles <tab> sample1idA <tab> sample1idB <tab> sample2idA <tab> sample2idB etc.
where sample1id is your identifier for the first sample, and the A, B correspond to the different allele intensities.
An example input intensity file is provided with the program for your information.
We recommend chunking the input data into separate files for each chromosome (each one in the format
above), and calling each chunk separately in parallel on a different processing node to significantly reduce execution time.
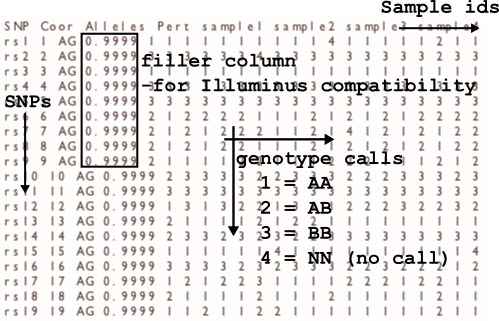
-out OUTPUT_FILE
The output file name, including the path. Two files will be created by the algorithm with the filepath specified. One will have the suffix '.calls' appended to it
for the genotype calls, and the other '.probs' for the posterior probabilities.
The calls file
The output format is space-delimited with columns: rs, coordinate, allelesAB, pertubation value, call_1, call_2, call_3,.... The order of the calls is given by
the order of the sample ids in the header line of the output file.
The calls are encoded as 1 = AA, 2 = AB (heterozygote), 3 = BB, 4 = NN (no call). The pertubation value is merely output to make call files more compatible with
those of the Illuminus caller, and not reflective of any pertubation analysis.
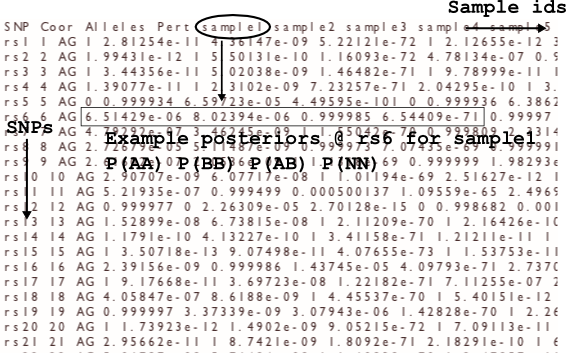
The probs file
The probs file gives probabilities for the samples in the header. For each sample at each SNP there are four probabilities, in the order: P(AA) P(BB) P(AB) P(NN). In cases where the maximum genotype probability is less than the probability threshold, the call will be NN but the posterior probabilities might not have P(NN) as the highest value.
OUTLIER HANDLING:
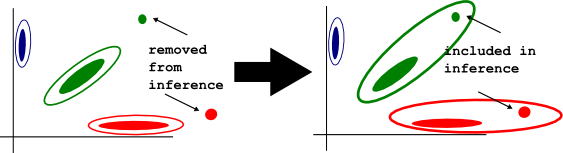
-nointcutoff
By default, when fitting the genotype mixture model, optiCall doesn't consider samples with outlying intensity values. They are still called once the mixture model has been fit. This option stops optiCall excluding such outliers from model fitting. Use this if you already have filtered samples/SNPs for intensity outliers.
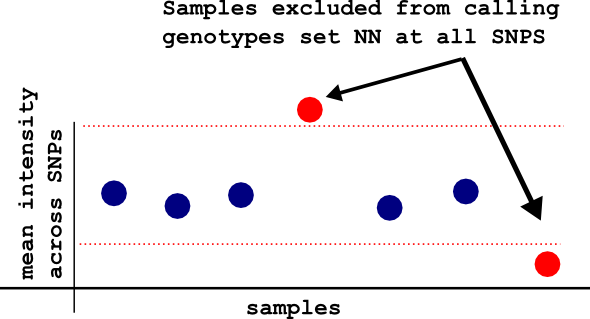
-meanintfilter
By running this flag, optiCall identifies samples with mean intensity values (across SNPs) that are too high or too low, and excludes them from the model fitting. This samples have their genotypes set to NN at all SNPs. If you already deal with outlying intensities before using optiCall, there's no need to use this flag.
-blank
This option means optiCall calls all samples at a SNP as NN if the best attempt at genotype clustering produces a Hardy-Weinberg Equilibrium (HWE) p-value of less than optiCall's threshold (by default 1e-15). By default optiCall will not blank SNP calls out of HWE.
PROVIDING SAMPLE INFORMATION
& CALLING A SUBSET OF SAMPLES:
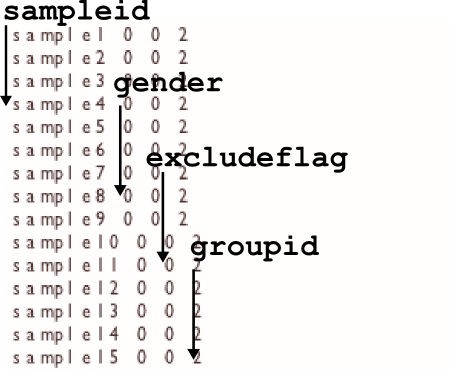
-info FILE
The info file gives optiCall extra information about the intensity data to help it make better calls. Use the file to specify sample genders (used in X, Y chromosome calling) and put samples into ethnicity based groups (to better calculate HWE p-values).
In addition the file can be used to exclude samples from genotype calling if you're only interested in calls from a subset of samples. It is whitespace separated and the format is:
It is whitespace separated and the format is:
sampleid <space> gender <space> excludeflag <space> groupid
with a line for all the samples in the intensity data supplied. All fields need to be supplied for each sample (though there are unknown values for each field, see table below). An example info file is provided with the optiCall download.
| field | type | description |
|---|---|---|
| sampleid | string | sampleid should match the sampleid given in the header of the intensity file. |
| gender | integer | gender is either 1 for male or 2 for female - and any other integer value is considered as unknown. |
| excludeflag | integer | excludeflag is 1 if the sample is to be excluded from calling, or zero if it is to be included in calling. |
| groupid | integer | The groupid is designed to account for possible ethnicity heterogeneity. For example when optiCall calculates HWE p-values, calling different ethnicities together could pose a problem. To handle this, give a separate groupid to each unique group being called. groupids are integers greater than or equal to 0, and an id of -9 will exclude the sample from any HWE calculations. |
CURATED PRIORS:
-inblock PRIOR_INTENSITY_FILE
optiCall begins by sampling intensities at random from the input file. These random intensities are clustered to form prior information of where each genotype's intensities are expected to cluster. The prior information is then used in the genotyping at each SNP. However, occasionally it may be undesireable to make a prior from the SNPs in the input intensity file. A lot of the SNPs may be expected to be of poor quality, or the file may contain overwhelmingly rare variants. Having only rare variants in the intensity file means optiCall can't create a good prior cluster for heterozygotes, resulting in impaired calling of hets.
In the event of these cases use the -inblock option, followed by an optiCall format intensity file, containing good quality SNPs, both rare and common (50:50 mix works well). Note the samples in the PRIOR_INTENSITY_FILE need to be the same and in the same order as those in the -in file. Also, any samples excluded from calling by an -info file, will also be automatically excluded by optiCall from the -inblock clustering.
X & Y CHROMOSOMES:
-X, -Y & -MT
Use these options when calling intensity files for X chromosome, Y chromosome and mitochondrial SNPs respectively.
For X and Y chromosomes the best calls are achieved by also using the -info option and supplying gender information for samples.
ADJUSTING THRESHOLDS:
-hwep NEW_VALUE
By default this is 1e-15. This is the threshold Hardy-Weinberg Equilibrium (HWE) p-value at which optiCall determines a clustering attempt has failed. If the HWE p-value is less than this threshold optiCall will first attempt a second rescue clustering for the SNP.
To get the best calls, we recommend you set this value to the HWE p-value QC threshold for your study, so that any SNPs that may be potentially lost could be fixed by the rescue clustering. If you're not sure about your QC thresholds, or want faster execution, the default will suffice.
-minp NEW_VALUE
By default this is 0.7. This is the threshold at which optiCall will make a call. If no posterior genotype probability is above this value, then optiCall sets the genotype to NN.
BEST PRACTICES:
Multiple Chromosomes
We recommend chunking the input data into separate intensity files for each chromosome (each one in the format here), and calling each chunk separately in parallel on a different processing node to significantly reduce execution time. For X, Y and mitochondrial files use the -X, -Y & -MT flags respectively.
Multiple Batches
If you have data from multiple genotyping centres, or in different batches (taken at different timepoints for example), we've found it is best (especially in terms of computational resources) to run optiCall on each batch in parallel instead of combining all the batches into one big intensity file.
Multiple Ethnicities
If a single batch contains multiple ethnicities, optiCall all ethnicities together, and provide ethinicity information using the -info option and file.
All of the above
Call each batch separately, and within each batch use the -info option and file to tell optiCall about the different ethnicities in the batch. For each batch, chunk the batch intensity files into chromosomes and run each in parallel (using the -X, -Y, -MT options for X, Y chromosomes and mitochondrial SNPs).
Custom arrays with a large sample size and overwhelmingly rare SNPs
In cases where an array is mostly composed of rare SNPs (e.g. chips with >70% rare SNPs such as the ExomePlus), and combined with a large sample size (> 10K samples, though exact number will vary), optiCall's prior clustering step may not sample enough heterozygotes to establish a good prior for the location and size of the heterozygous cloud. You can see this by looking at the priors optiCall prints out. The "mus" output are the (x,y) locations of the centre of the genotype clusters. They should correspond to two homozygous clusters (large x, small y and small x, large y), one in between - the het cluster (x and y have similar values), and a (0,0) cluster to catch outliers. If this is not happening, it may be the chip has an abundance of rare SNPs. Should this be the case, create a separate intensity file, containing a 50:50 mix of rare and common good quality SNPs. This file can be fed to optiCall using the -inblock option described above.
STUDIES USING OPTICALL:
Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis.
International Multiple Sclerosis Genetics Consortium (IMSGC), Nat Genet. 2013 Nov;45(11):1353-60. doi: 10.1038/ng.2770. Epub 2013 Sep 29.
Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway.
Lee JC et al., Cell. 2013 Sep 26;155(1):57-69. doi: 10.1016/j.cell.2013.08.034. Epub 2013 Sep 12.
Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci.
International Genetics of Ankylosing Spondylitis Consortium (IGAS), Nat Genet. 2013 Jul;45(7):730-8. doi: 10.1038/ng.2667. Epub 2013 Jun 9.
Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis.
Liu JZ et al., Nat Genet. 2013 Jun;45(6):670-5. doi: 10.1038/ng.2616. Epub 2013 Apr 21.
Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease.
Jostins et al., Nature. 2012 Nov 1;491(7422):119-24. doi: 10.1038/nature11582.
Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis.
Liu JZ et al., Nat Genet. 2012 Oct;44(10):1137-41. doi: 10.1038/ng.2395. Epub 2012 Sep 9.